
3a) Scaling and analysing datasets
f' and f'' for Se varies with wavelength as (output from program crossec):
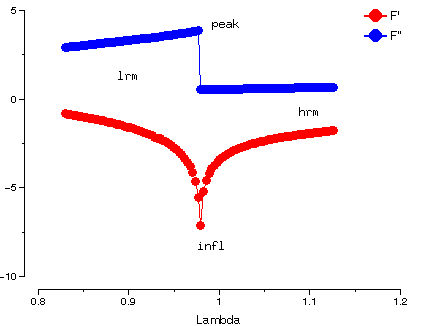
We have data for 4 wavelengths, labelled as:
- lrm
- Low wavelength remote - f' small, f'' large
- peak
- Peak of absorption - f' large, f'' very large
- infl
- Point of inflection - f'' very large, f'' small
- hrm
- High wavelength remote - f' small, f'' very small
But:
- Need to check data is labelled properly
- "Peak" may not be exactly on peak, so check real strength of anomalous signal
Normal Probability plots
See Lynne Howell and Dave Smith, J.Appl. Cryst. 25 81-86 (1992)3b) Preparing datasets for finding heavy atoms
Normalised Structure Factors
Most direct methods procedures make use of normalised structure factors (denoted E) rather than the bare structure factor amplitude F. The value of E for a reflection is defined as F divided by the product of epsilon (a factor dependent on the Laue group symmetry) and the r.m.s. value of the structure amplitudes at its sin(theta)/lambda value. The values of E therefore do not fall off with increasing scattering angle.See C.Giacovazzo et al, Fundamentals of Crystallography, p.321
In CCP4, the program ECALC is used to derive Es from Fs. These can then be used in the direct methods program RANTAN.
3c) Find heavy atoms
Heavy atom positions
When trying to understand heavy atom positions, remember to consider symmetry equivalent positions. Also, depending on the spacegroup, there may be alternative origins. Finally, there are 2 possible hands for each set of positions.The current example is in spacegroup C2. This is a polar spacegroup, so that the origin is not fixed along the b axis. In addition, there are 4 possible origins in the a-c plane:
Norigin Xo Yo Zo
1 0.0000 0.0000 0.0000
2 0.0000 0.0000 0.5000
3 0.5000 0.0000 0.0000
4 0.5000 0.0000 0.5000
Heavy atom sites from different phase sets output from RANTAN may be with respect to different origins. For example, the first 3 sites from phase set 1 are:
0.26 0.06 0.75
0.43 0.24 0.38
0.20 0.45 0.36
The opposite hand would also be a solution:
-0.26 -0.06 -0.75
-0.43 -0.24 -0.38
-0.20 -0.45 -0.36
We can then change the origin to -0.5,-0.24,-0.5 (origin 4 above, plus a shift along the b axis):
0.24 0.18 0.75
0.07 0.00 0.12
0.30 0.79 0.14
Finally, we find a symmetry mate of site 3 by applying the symmetry operation 1/2-X,1/2+Y,-Z:
0.24 0.18 0.75
0.07 0.00 0.12
0.20 0.29 0.86
These are in fact the first 3 sites of phase set 2!!
3d) Heavy atom refinement
Describe Derivatives and Refinement
In MAD, the "derivatives" correspond to different wavelengths of the same derivative (e.g. a 3 wavelength Se-Met MAD experiment would give 3 "derivatives"). When refining heavy atom positions for each "derivative", you are actually refining the same heavy atom coordinates (e.g. Se coordinates) against different data for the different wavelengths.For each heavy atom, you can refine its XYZ coordinates, its occupancy and its B factor. For each "derivative" or wavelength, you can refine the heavy atom parameters against:
- isomorphous data
- the difference in the average structure factor F at that wavelength and the F at a reference wavelength (the "native", usually chosen to be the inflection point wavelength)
- anomalous data
- the anomalous difference D at that wavelength